What’s the FDA up to on Patient-Focused Drug Development (PFDD) these days?

Part of the FDA’s mandate in implementing the “21st Century Cures Act” is ensuring the incorporation of patient experience data into drug development. They’ve hit the deadline for another Patient Focused Drug Development (PFDD) Guidance document, so let’s look at the status.
Background
The Agency is in the process of issuing four PFDD guidance documents to “address, in a stepwise manner, how stakeholders can collect and submit patient experience data” for medical product development and regulatory decision-making.1 Draft Guidance 1 on “Collecting Comprehensive and Representative Input”2 came out last June, and Draft Guidance 2, “Methods for Identifying What is Important to Patients,”2 was due out at the end of Q2 this year, according to the Plan3 — but hasn’t been released yet.
There’s encouragement; at last July’s FDA-AACR Real World Evidence workshop, Jacqueline Corrigan-Curay, CDER’s Director of the Office of Medical Policy, presented multiple examples4 of the FDA accepting real world data and real world evidence (RWD/RWE) when it approved new indications of previously approved drugs, one of which was from 1998.
But it leaves the industry in the same position – with a handful of examples of the kind of data and format the FDA considered acceptable.
Latest issuance
In May, the FDA issued a document on that crucial subset of PFDD: Submitting Documents Using Real World Data and Real World Evidence (RWD/RWE) to FDA for Drugs and Biologics: Guidance for Industry , “…to encourage sponsors and applicants who are using real-world data (RWD) to generate real-world evidence (RWE) as part of a regulatory submission to FDA to provide information on their use of RWE in a simple, uniform format.”5 At first glance, it’s underwhelming – the uniform format they’re recommending is using a cover sheet on submissions to indicate there’s RWD/RWE inside. There’s an example of one in the Appendix.
However, the Guidance is significant. It indicates how the FDA initially plans to incorporate RWD into the approval process; what kind of product claims the RWE is expected to support; and what kinds of real world data the FDA anticipates receiving.
Internal Use Only: (IND, NDA, BLA)
The introductory paragraph says that once the cover letter draws their attention to the fact that there’s RWD/RWE in the submission, the information is:
- Only going to be used “for internal tracking purposes”
- Only tracked for three kinds of submissions: investigational new drug applications (INDs), new drug applications (NDAs), and biologics license applications (BLAs)
- Only as supporting evidence for regulatory decisions regarding safety and/or effectiveness
In contrast, the FDA doesn’t plan to track submissions that are not tied to a specific product or that are not supporting a regulatory decision about safety or effectiveness, such as natural history studies to develop a biomarker, feasibility studies, and exploratory analyses.
Examples of RWD
Furthermore, the FDA defines what it currently considers RWD: information about patient health status and health care delivery that are “routinely collected from a variety of sources.” Describing the sources as “examples,” they listed:
- Data derived from electronic health records (EHRs)
- Medical claims and billing data
- Data from product and disease registries
- Patient-generated data, including in-home use and/or other decentralized settings
- Data gathered from other sources that can inform on health status, such as mobile devices
The guidance clearly opens the gate to patient-generated data – like online postings, interviews and survey results – data generated outside the clinical trial setting, and to mobile devices.
Getting from Data to Evidence: Analytics
Because they define RWE simply as, “the clinical evidence regarding the usage and potential benefits or risks of a medical product derived from analysis of RWD,” the examples are helpful here, too: “[such as] collecting information about effectiveness or safety outcomes from an RWD source in randomized clinical trials or in observational studies.” The evidence can only be about effectiveness or safety, and they’re expecting it from data gathered in the established settings of clinical trials and observational studies, with the evidence derived via “evolving analytic techniques.”
The Cover Letter: Specify Purpose, Study Design, and Source
The meat of the Guidance is in the appendix, because it establishes that the first thing they want to see in the cover letter is the “purpose(s) of using the RWE in the submission,” and the checklist itemizes the expectations. Data can be used:
- To provide evidence in support of effectiveness or safety for a new product approval
- To provide evidence in of support labeling changes for an approved drug, including:
- Add or modify an indication
- Change in dose, dose regimen, or route of administration
- Use in a new population
- Add comparative effectiveness information
- Add safety information
- Other labeling change. Specify:
- To be used as part of a post-marketing requirement to support a regulatory decision
They expect this evidence to be derived from studies using established designs, but leave it open:
- Randomized clinical trial
- Single arm trial
- Observational study
- Other study design. Specify:
And finally, there’s a checklist of the kinds of RWD already detailed in this article.
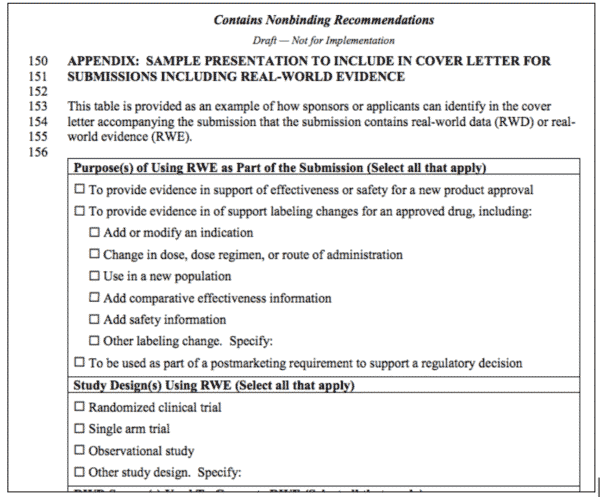
Figure 1: Excerpt from FDA Guidance for Industry, “Submitting documents using real world data and real world evidence to FDA for drugs and biologics,” May 2019
Industry Commentary
There are only 9 submissions to the archive of public commentary on the release, but they include commentary from Gilead and Novartis, as well as from the trade organizations PhRMA, BIO, and ACRO. What are these players looking for? Among other things, they’ve asked for:
- Gilead: adding supplemental new drug applications (sNDAs) and supplemental biologics license applications (sBLAs) to the list of acceptable submissions
- Novartis: adding RWD/E from pharmacy claims to inform regulatory decisions and expanding the defined scope to include other forms of “routinely” collected data such as Patient reported outcomes (PROs) and quality of life (QALY) metrics
- PhRMA: a standard process and harmonizing submission tracking across different centers in the FDA, including CDRH, CDER, and CBER.
- BIO: the FDA to support its call for RWD/E with a robust and transparent infrastructure to handle it, along with a better idea of what the Agency is working with, such as “aggregate, de-identified data on the number of RWE submissions made, context-of-use, study design, data type, and therapeutic area” so that stakeholders know what the Agency is working with
- ARCO: publicly available information such as “standardized summary assessment of the quality of data underlying RWD submissions”
Finally, more than one comment observed that, while the Draft Guidance presents definitions for the terms RWD and RWE, there are instances in the same document where it seems to be using both terms inconsistently. That’s indicative of the larger issue: the Agency is clearer about the goal – making drug development patient-centric – than it is about the infrastructure and the process, even down to settling on the use of basic terms.
Stakeholders want clearer parameters on the approval process and what constitutes acceptable data. The FDA is trying to articulate the parts of what will be a monumental change in its drug approval process. Perhaps it continues to be an issue of, “I don’t know what it is that I want, but I’ll know it when I see it.” They are setting some basic boundaries and looking for examples of how companies might implement them in the context of today’s submissions.

Inspire offers a trusted community to patients and caregivers. Our goal with this blog, this website and our content is to provide the life science industry access to the true, authentic patient voice. In so doing, we support faithful operationalization of patient-centricity. Take a look at our case studies, eBooks and news outlet coverage.
References
1 https://www.fda.gov/drugs/development-approval-process-drugs/fda-patient-focused-drug-development-guidance-series-enhancing-incorporation-patients-voice-medical
2 https://www.fda.gov/media/113653/download
3 Plan for Issuance of Patient Focused Drug Development Guidance. May 2017. https://www.fda.gov/media/105979/download
4 https://www.aacr.org/AdvocacyPolicy/GovernmentAffairs/Documents/FDA-AACR_RWE_SlideDeck.pdf, Slide 9.
5https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submitting-documents-using-real-world-data-and-real-world-evidence-fda-drugs-and-biologics-guidance
6 ibid.